 Dr. Gillies Lab
Dr. Gillies Lab
Targeting Tumor Metabolism to Enhance Immune Therapy
Project Summary:
The extracellular pH (pHe) of solid tumors is unequivocally acidic. We have generated strong preliminary evidence that tumor acidity inhibits anti-tumor immunity by suppressing the activation of T-cells. Importantly, neutralizing this acidosis with oral buffer therapy synergizes with anti-tumor immune therapies, such as checkpoint (anti-CTLA4, anti-PD1) blockades, and adoptive T-cell therapy. It is now accepted science that activation of CD8+ T-cells with antigen leads to an increased rate of aerobic glycolysis, and that this is required for T-cells to carry out their cytokine release and cytotoxic functions. We (1), and others (2, 3) have shown that placing T-cells at low pH (6.7), even after 24-48 hr of activation with antigen, reverts them to a more oxidative metabolic phenotype, abrogates their cytokine release, and inhibits their cytotoxic activity. These effects are robust, and have been consistently observed with T-cells from pmel mice activated with gp100, OVA-II mice activated with SIINFEKL, and native T-cells from mice and humans activated with anti CD3/CD28 with identical results. Critically, following 24 hr at acid pH, cells retain their ability to secrete IFNγ when re-incubated at pH 7.4. However, the mechanism by which acid pH inhibits T-cell function is not known. We know it is not due to an effect on intracellular pH (1). Hence our project is propose that the effect is mediated by one (or more) acid-sensing G-proteins or ion channels that have been identified over the last decade (4).
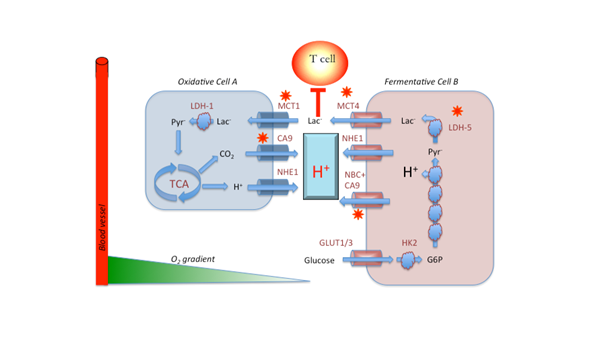
Metabolic symbiosis in cancer, after Sonveaux. Oxidative cells (A) consume lactic acid, which is converted to pyruvate and subsequently oxidized in the TCA cycle to produce ATP and CO2. The CO2 is hydrated on the outside of the cell by carbonic anhydrases 9 or 12 (CA9/12) to produce HCO3- + H+. Additional H+ are exported by the sodium/hydrogen exchanger, NHE. In fermentative cells (B), glucose is imported via GLUT-1 or GLUT-3, converted to G6P, whereupon it enters glycolysis. Notably, the oxidative step catalyzed by GAPDH results in acid production and release of the H+. These H+ are removed by NHE1 and the sodium dependent bicarbonate transporter, NBC, working in concert with CA9. The lactate produced by reduction of pyr to lactate by LDH-5 (a.k.a. LDH-A) is exported by MCT4.
We are investigating pharmacological alternatives to buffer therapy, based on known pathways to affect tumor acid-base balance (see graphical abstract). In preliminary screens, we have identified two targets (carbonic anhydrase IX and LDH-A) that were inhibited with first-generation inhibitors, DH348 and FX-11, respectively. In an orthotopic Panc02 model, DH348 by itself was inhibitory to tumor growth . Notably, FX11 had no effect as monotherapy, but appeared to synergize with anti-PD-1 response. Two second-generation LDH-A inhibitors (006 and 737) have been obtained from the NIH, and 737 also shows additive effects to anti-PD1 therapy in this same system.
References:
- Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, Damaghi M, Wojtkowiak JW, Mule JJ, Ibrahim-Hashim A, Gillies RJ. Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy. Cancer research. 2016;76(6):1381-90. doi: 10.1158/0008-5472.CAN-15-1743. PubMed PMID: 26719539; PubMed Central PMCID: PMC4829106.
- Lardner A. The effects of extracellular pH on immune function. Journal of leukocyte biology. 2001;69(4):522-30. PubMed PMID: 11310837.
- Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, Cova A, Canese R, Jachetti E, Rossetti M, Huber V, Parmiani G, Generoso L, Santinami M, Borghi M, Fais S, Bellone M, Rivoltini L. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer research. 2012;72(11):2746-56. doi: 10.1158/0008-5472.CAN-11-1272. PubMed PMID: 22593198.
- Damaghi M, Wojtkowiak JW, Gillies RJ. pH sensing and regulation in cancer. Frontiers in physiology. 2013;4:370. doi: 10.3389/fphys.2013.00370. PubMed PMID: 24381558; PubMed Central PMCID: PMC3865727.
- Ibrahim Hashim A, Cornnell HH, Coelho Ribeiro Mde L, Abrahams D, Cunningham J, Lloyd M, Martinez GV, Gatenby RA, Gillies RJ. Reduction of metastasis using a non-volatile buffer. Clinical & experimental metastasis. 2011;28(8):841-9. doi: 10.1007/s10585-011-9415-7. PubMed PMID: 21861189; PubMed Central PMCID: PMC3213349.
- Ibrahim Hashim A, Wojtkowiak JW, Ribeiro M, Estrella V, Bailey KM, Cornnell HH, Gatenby RA, Gillies RJ. Free Base Lysine Increases Survival and Reduces Metastasis in Prostate Cancer Model. J Cancer Sci Therap. 2011;2011:S1-S4.
- Ibrahim-Hashim A, Cornnell HH, Abrahams D, Lloyd M, Bui M, Gillies RJ, Gatenby RA. Systemic buffers inhibit carcinogenesis in TRAMP mice. J Urol. 2012;188(2):624-31. Epub 2012/06/19. doi: 10.1016/j.juro.2012.03.113. PubMed PMID: 22704445; PubMed Central PMCID: PMC3694604.
Project Members
Arig Ibrahim-Hashim, MD
Research Scientist