 Ruffell Lab
Ruffell Lab
Ruffell Lab
Ruffell Lab
General considerations: To avoid artifact issues due to non-specific targeting it is recommended to pick at least 3-5 independent “single guide RNA” (sgRNA) sequences for each gene of interest or non-targeting control. Once the knockout efficiency is verified, choose the best 2-3 gRNA knockout cell lines that will be used to study the knockout of your gene of interest. Note: If non-specific targeting seems to cause issues in your system, the non-specific genes targeted can be determined via their sequence similarity. Simply enter the target sequence on Nucleotide Blast to find genes with similar sequences:
Note: 5 considerations for target site selection. Knockout libraries often target 5′ constitutively expressed exons, but activation and repression libraries will target promoter or enhancer regions. For gene knockout it is recommended to choose early constitutively expressed exons but try to avoid the first exon if not possible otherwise. IDT Integrated DNA Technology provides a detailed summary of considerations. Here in short:
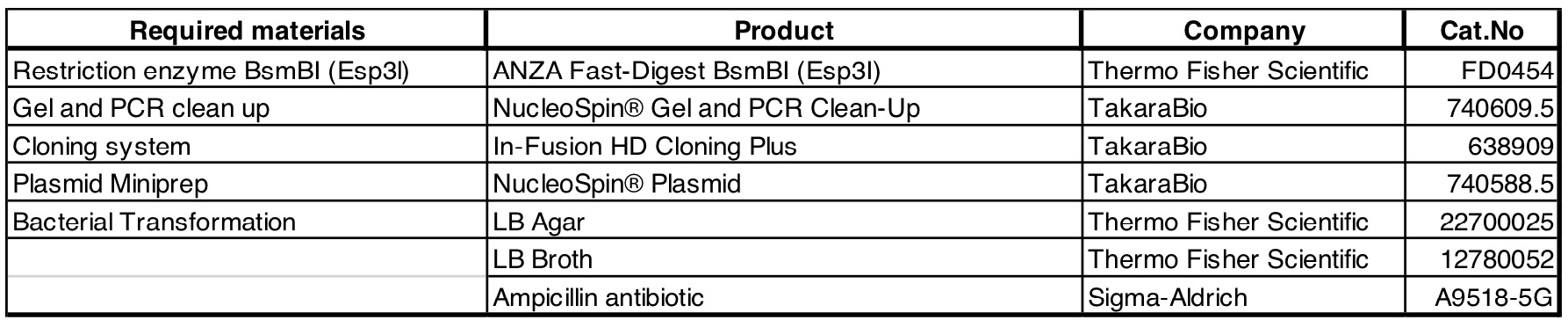
Required materials:
General considerations: Choose the plasmid system depending on the characteristics of the target cells. Either use a 1-plasmid system that includes the Cas9 on the sgRNA-containing plasmid, or a 2-plasmid system in which Cas9 must be delivered separately. Due to the large size of Cas9 (4.1 kbp) and depending on the characteristics of the target cell type it may be better to use the 2-plasmid systems over a 1-plasmid system due to the fact that lentiviral titers decrease with increased size of inserts. This becomes important for difficult-to-infect cells such as primary cells, while 1-plasmid systems are more convenient for easy-to-infect cells as it also reduces the number of selection markers being integrated. There are several plasmid systems available from addgene.org as described in the section “Select expression system” of the CRISPR guide.
A variety of different 1- or 2-plasmid systems for expression in a variety of species are available on addgene.org. Information and availability of empty CRISPR plasmids can be found here.

Schematic overview of DNA oligo design and cloning reaction for single sgRNA cloning

NOTE: The reaction mix can been downscaled 4-5x from the manufacture’s recommendations in order to save In-Fusion HD Enzyme Premix. If the concentration of the linearized vector is too low it needs to be upscaled accordingly. If recovery of linearized plasmid is too low from the PCR/Gel cleanup, try to improve recovery by multiple elution steps (see your manufacture’s recommendations) or upscale initial amount of vector for digest.
5. Transformation of competent bacteria using Stellar™ competent cells. Make sure bacteria are thawed on ice 10-15min before using. Transfer 50μl of bacteria into a pre-cooled 14ml round-bottom tube (Falcon tube; Corning 352059), add 2.5μl of the In-Fusion cloning reaction and place on ice for 30min. NOTE: Stellar™ competent cells are recommended and delivered with the In-Fusion HD cloning kit, however other competent bacteria strains can be used alternatively.
6. Heat shock the cells for exactly 45 sec at 42°C and place back on ice for 1-2min.
7. Add 450μl of pre-warmed (37°C) SOC medium to make a total volume of 500μl and incubate with shaking (160-225 rpm) for 1h at 37°C.
8. Transfer 1/5 – 1/3 of each transformation into a separate microfuge tube and after spinning at 6000 rpm for 5 min reconstitute with 100 μl fresh SOC medium and plate on a 37°C pre-warmed LB-agar plate containing 100 μg/ml of ampicillin. Incubate plates at 37°C overnight.
9. The next day, pick 3-5 single colonies from each plate and set up a 3-5ml liquid LB culture overnight at 37°C to isolate DNA the next day via your standard method of choice (e.g. Miniprep/Midiprep). To determine the presence of inserts, analyze the DNA by restriction enzyme digest, PCR screening or preferentially sequence the insert (use U6 forward primer).
10. Expected results:
11. Once positive clones are confirmed proceed to produce lentivirus and generate sgRNA/Cas9 mediated stable gene knockout cell lines either using your standard method of choice or proceed as outlined below.
General considerations: For difficult to infect cells such as primary cells, it may require concentrating lentiviral supernatant via ultracentrifugation at 24’000 – 26’000 rpm for 2h at 4°C using an ultracentrifuge with swinging bucket. Preferentially fresh lentiviral supernatant should be used for infection, but it can be frozen at -80°C for up to 1 year.
{kind=link}