 E. Schönbrunn Laboratory
E. Schönbrunn Laboratory
E. Schönbrunn Laboratory
E. Schönbrunn Laboratory

Research from the Ernst Schönbrunn laboratory focuses on the elucidation of the structure-activity relationship of medicinally important proteins. We use protein crystallography and cryo-electron microscopy combined with methods in biochemistry and cell biology to explore protein-inhibitor and protein-protein complexes at the atomic level. Our goal is to facilitate the discovery and development of new inhibitors as potential therapeutics.
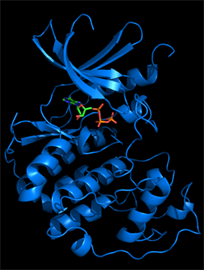
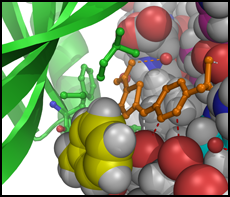
We typically follow two routes towards the discovery of new inhibitors, which interconnect if the 3D structure of a target protein-inhibitor complex can be determined. The first route is an empirical approach involving the development of an assay suitable for the screening of compound libraries for binding or inhibitory activity. Discovered ligands/inhibitors (hits) will be scrutinized by structure-activity relationship (SAR) studies including biochemical and cell-based methods until the most efficacious inhibitors with drug-like properties (leads) have been identified. The second route is the rational design of inhibitors based on the 3D atomic structure of the target protein. Our method of choice is protein crystallography to determine high-resolution structures of the target protein in the presence of newly discovered and known inhibitors or cofactors and substrates. For drug targets that are difficult to crystallize we attempt structure determination by cryo-electron microscopy. With structural information in hand, we perform computational studies (in silico design), such as molecular docking, to design compounds that satisfy the criteria for drug efficacy in cancer models and good pharmacological properties for clinical translation. We closely collaborate with medicinal chemists, cancer biologists and clinicians to facilitate the translation from early lead compounds to mature drug candidates.